

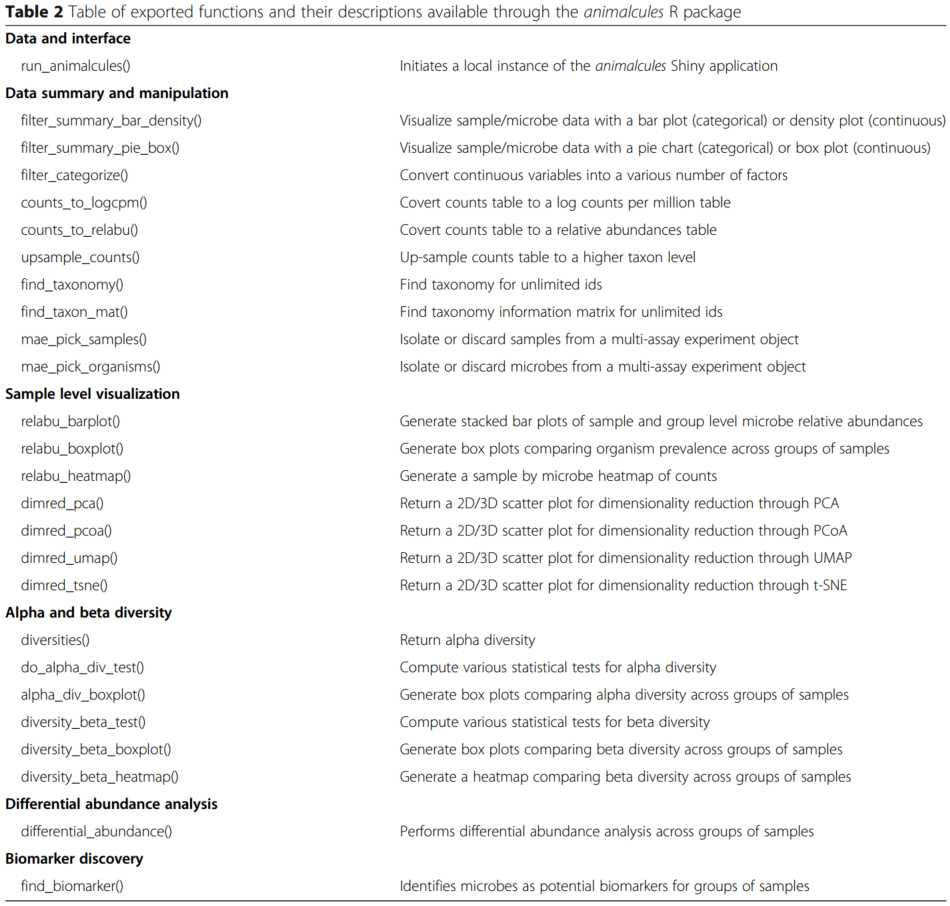
在右侧面板中,显示了连续变量的数据汇总指标表、散点/箱线图和密度图。对于分类变量,animalcules将自动识别并显示饼图和条形图。
数据可视化
一个典型的分析涉及可视化样品或样品组中微生物的丰度。animalcules实现了三种常见的可视化图类型,包括堆叠的条形图,热图和箱形图。使用animalcules :: relabu_barplot()生成的堆叠条形图用于可视化每个样品中给定分类水平上微生物的相对丰度,以单个条形图表示(图2)。条形图可以通过一个或多个样本属性进行颜色标记,还可以通过汇总组内的微生物丰度,通过这些属性来汇总样本。这是研究人员在不同的物种分类水平上识别样本或类群水平模式的有效方法。用户还可以选择根据样本属性或一个或多个生物体的丰度对条形图进行排序。还有一种简便的分离或去除样品的方法。使用该工具,用户可以快速浏览样本属性和物种分类级别的不同组合,以发现一个或多个组中的差异丰度,群落概况方面的离群值以及未被已知属性表示的样本群。
图 2 animalcules 丰度选项卡
animalcules Abundance tab
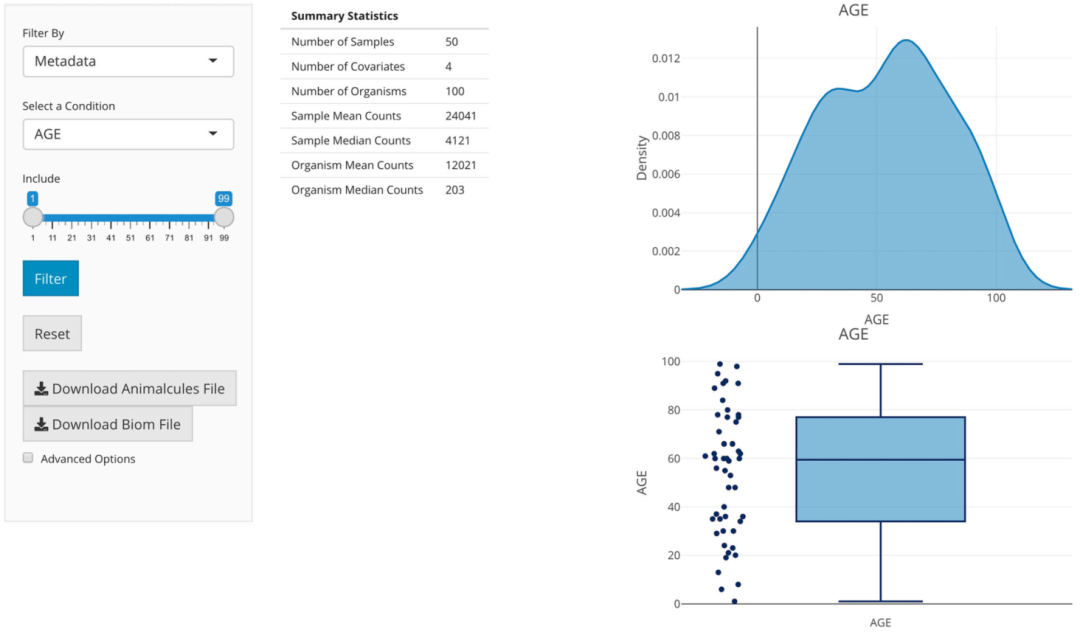
在子选项卡面板中,用户可以在α多样性分析和β多样性分析之间进行选择。在beta多样性分析中,右侧面板控制着要使用的统计检验,要检验的条件,并在表格和箱线图中显示统计检验结果。
降维
在任何数据分析工作流中,一个关键的步骤是在一个低维空间中可视化和汇总高变数据(图4)。在animalcules中,我们实现了四种常用的降维技术,包括主成分分析(PCA),主坐标分析(PCoA),t-分布随机邻接Eebedding (t-SNE)和均匀流形近似和投影(UMAP)。PCA和PCoA都将样本投影到一组新的轴上,由此最大的变化量由第一,第二和第三轴解释,而t-SNE和UMAP是将数据映射到低维嵌入的非线性方法。数据集的降维值可以输出到MAE对象中,也可以作为单独的表或文件输出。
图 4 animalcules 降维选项卡
animalcules Dimension Reduction tab.
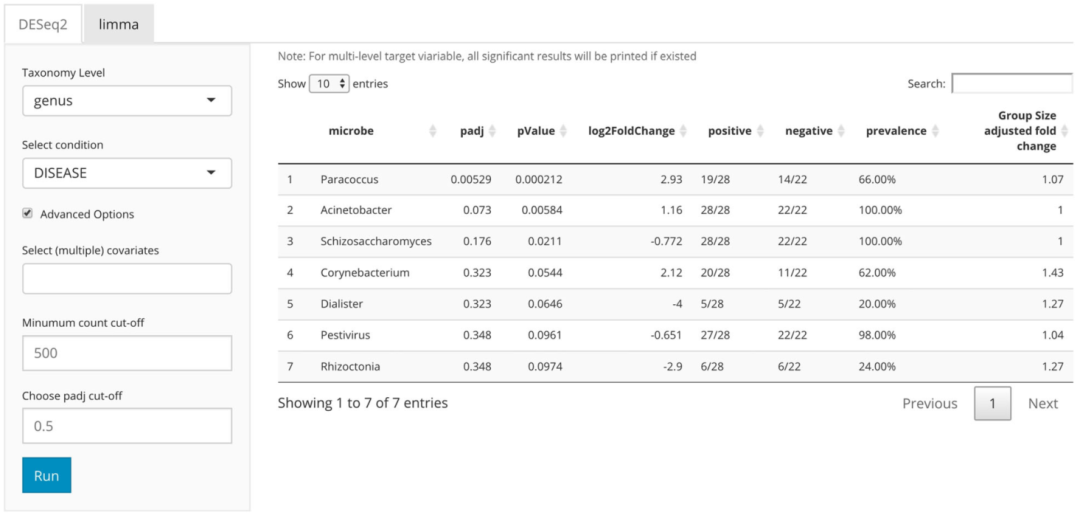
在子选项卡面板中,用户在DESeq2和limma之间进行选择。在左侧面板中,用户指定物种分类水平,目标条件,协变量,计数界限和调整后的p值阈值。在右侧面板中,显示了详细的差异丰度结果表。
生物标志物鉴定
animalcules的一个独特功能是生物标志物识别模块。用户可以选择逻辑回归或随机森林分类模型来识别微生物生物标志物。除了用于评估生物标志物预测性能的AUC值和平均交叉验证ROC曲线外,还将提供每个微生物的特征重要性评分(图6)。也可以通过命令行函数animalcules :: find_biomarker()进行生物标志物的识别。
图 6 animalcules 生物标志物选项卡
animalcules Biomarker tab
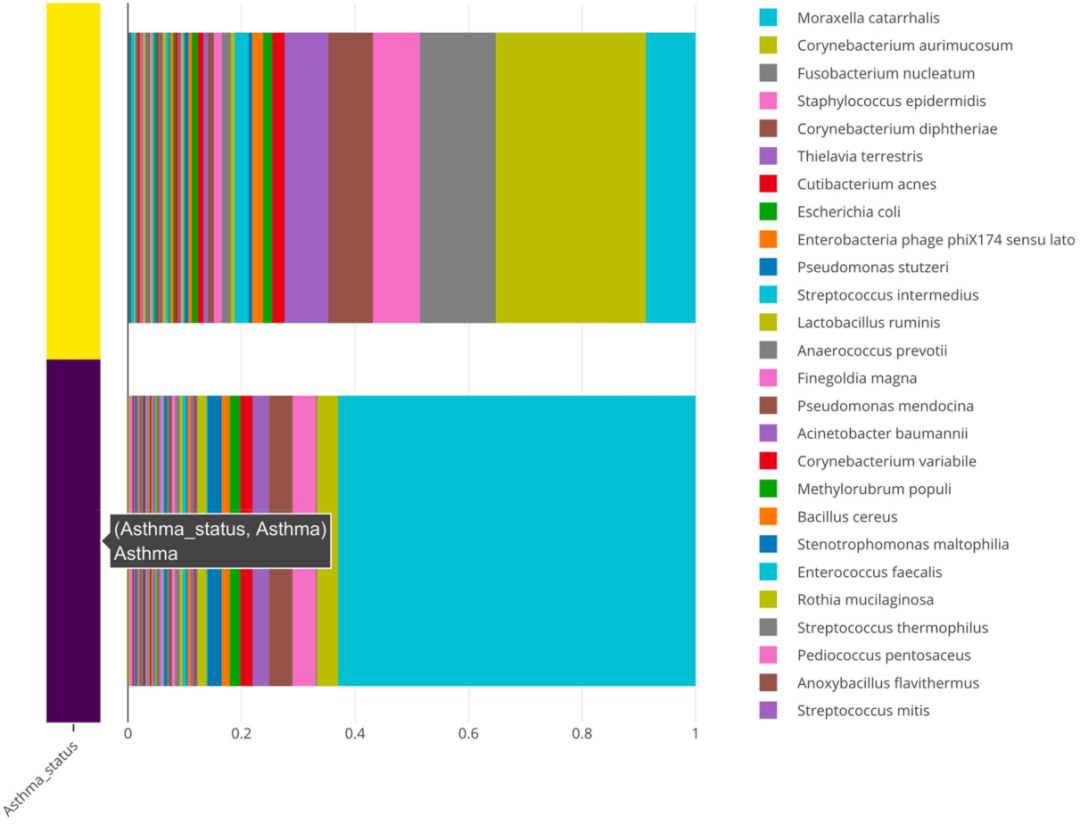
堆积的条形图,表示哮喘患者(紫色)和健康对照者(黄色)中微生物物种的群体相对丰富度。
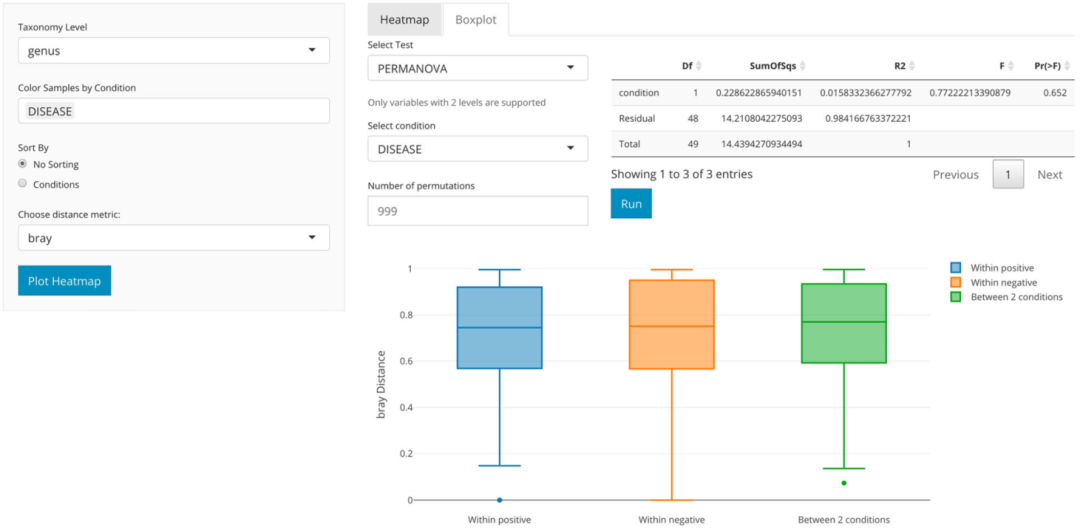
为了进一步研究哮喘患者和对照组中M. catarrhalis 和 C. aurimucosum的过度表达和表达不足,我们使用由animalcules :: boxplot()函数生成的箱线图可视化各组中的相对丰度并更好地了解各样本分布的平均值和方差。这些图通过显示丰度的巨大差异证实了先前的结果(图8)。此外,我们使用DESeq2对哮喘患者和对照组进行了微生物物种的差异丰度分析。该分析表明,M. catarrhalis在哮喘患者中的比例显著(q = 1.78e-3)过高(Log2FC = 5.9)。它还显示在对照中C. aurimucosum过高表达(Log2FC = 2.66),但并未达到统计显着水平(q = 0.236)。该表是使用animalcules :: differential_abundance()功能生成的。
图 8 差异丰富物种的相对丰度箱线图
Relative abundance boxplot for differentially abundant species
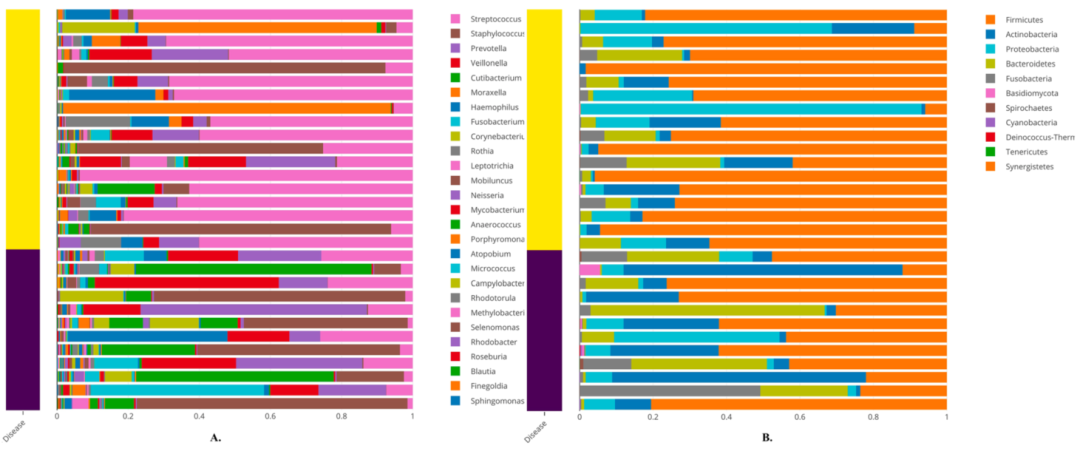
堆积条形图,表示结核菌(黄色)和健康对照(蓝色)样本中微生物菌种的样本方式相对丰度。子图a为属水平,子图b为门水平
从物种分类柱状图上,我们发现结核病样本与对照样本之间存在着不同的模式。在属水平上(图9a),与对照样品相比,结核病样品中的Streptococcus似乎具有较高的相对丰度。在门的水平上(图9b),我们发现在结核样本中厚壁菌门(Firmicutes)更为丰富。这两个图都是使用命令行函数animalcules :: rebalu_barplot()生成的。
为了定量了解结核病样本和对照样本之间的生态多样性差异,我们比较了样本的α和β多样性。对于alpha多样性,我们比较了结核样本和对照样本中的Shannon指数(见图10a)。animalcules 对这些多样性测度自动进行了非参数Wilcoxon秩和检验和参数Welch两样本T检验。此处,Wilcoxon秩和检验的p值为0.0060,而Welch两样本T检验的p值为0.0077,说明结核病组和对照组之间存在显著差异。从箱线图中,我们观察到对照组中的α多样性更高。alpha多样性箱图是由animalcules :: alpha_div_boxplot()生成的,而统计检验是由animalcules :: do_alpha_div_test()生成的。
图 10 结核病示例数据集多样性分析
TB example dataset diversity analysis
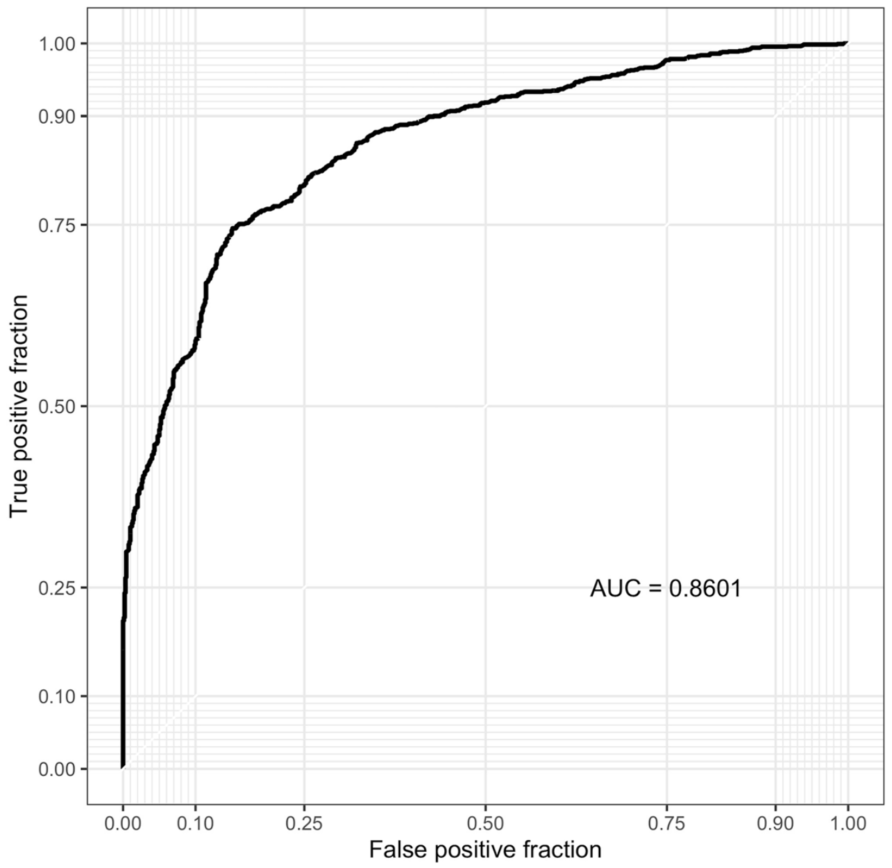
ROC显示了已识别生物标志物的AUC和交叉验证预测性能。
综上所述,在animalcules的帮助下,我们探索并比较了结核病样本和对照样本之间的微生物群落差异。我们的分析表明,与结核病组相比,对照组的微生物群落结构更加多样化,分布更均匀。
此外,结核病组和对照组均具有一个特定的在该组内共享的微生物组成。最后,我们确定了一个微生物子集,表明其在结核病与对照样品之间的区分能力,其可以用作新的结核病的生物标志物。
讨论
animalcules 的基本特征是其通过动态可视化工具与用户的无缝交互。这种设计逻辑基于这样一个事实,这种设计逻辑的根源在于微生物学研究人员必须在多个层次(物种分类法)和多个尺度(标准化)上对数据进行分析,如果没有一个有组织的分析框架和工作流程,数据可视化和分析将变得复杂。animalcules解决了这个问题,它提供了一个平台来交互式地探索大型数据集,使用户可以通过适当的分析方法更容易地识别数据集内在的模式。关键分析方法允许用户探究门水平上多个样本组之间的组间相对丰度模式的差异,检查一个特定样本组中最丰富的物种,或检查不同物种分类水平上的单个样本微生物组组成。可以通过alpha / beta多样性统计检验,差异丰度分析以及生物标志物识别来进一步检验识别出的模式。
此外,animalcules 利用MAE对象(一种用于多组测序数据的有效数据结构),在未来可以扩展到包含宿主测序分析,并使分析宿主-微生物相互作用的简洁方法成为可能。animalcules 的灵活性使其成为将来跨不同数据源和下游分析进行整合的有用工具:除了帮助分析16S rRNA和鸟枪测序微生物组数据外,animalcules 可以用来可视化模拟微生物组研究和涉及合成微生物群落的微宇宙实验的结果。通过animalcules 处理的物种分类学丰度信息可以进一步用于微生物共现网络的系统评估,扩大复杂数据集的信息可视化的可能性,并与微生物-微生物相互依赖的机制模型进行比较。
在可重现性方面,R/Shiny在本文发表时的一个不幸缺陷是缺乏一个用于生成历史记录或进行跟踪分析的系统结构。更具体地说,因为界面中的每一分钟的变化(例如,过滤数字尺度)都会触发应用程序服务器端的操作,所以记录R History可能会变得太大,太快而没有用处。相反,为了再现性,我们建议使用GUI进行初步和探索性分析,然后在R Markdown文档中使用命令行函数和代码创建可共享的再现性报告。
结论
在这份报告中,我们介绍了*animalcules– *一个开源的R包和Shiny应用程序,致力于16S rRNA和鸟枪测序(宏基因组学和宏转录组学)数据的微生物组分析。我们将领先和新颖的方法纳入一个高效的框架中,以使研究人员能够描述和理解其数据中的微生物群落结构,从而对微生物群落和感兴趣的表型之间的联系提供有价值的见解。
可用性和要求
项目名称:animalcules
项目主页:https://github.com/compbiomed/animalcules
官方/正式发布:https://www.bioconductor.org/packages/release/bioc/html/animalcules.html
帮助文档:https://compbiomed.github.io/animalcules-docs/
操作系统:Linux, OS X, Windows
编程语言:R
许可证:GNU GPLv3
数据和资料可用
Animalcules可以在GitHub (https://github.com/compbiomed/animalcules)或Bioconductor (https://bioconductor.org/packages/release/bioc/html/animalcules.html)上免费获得,并在 https://compbiomed.github.io/animalcules-docs/
上附有完整的文档和教程。
Reference
Yue Zhao,Anthony Federico,Tyler Faits,Solaiappan Manimaran,Daniel Segrè,Stefano Monti,W. Evan Johnson. animalcules: interactive microbiome analytics and visualization in R.Microbiome, (2021) 9:76
https://doi.org/10.1186/s40168-021-01013-0
猜你喜欢
10000+:菌群分析 宝宝与猫狗 梅毒狂想曲 提DNA发Nature Cell专刊 肠道指挥大脑
系列教程:微生物组入门 Biostar 微生物组 宏基因组
专业技能:学术图表 高分文章 生信宝典 不可或缺的人
一文读懂:宏基因组 寄生虫益处 进化树
必备技能:提问 搜索 Endnote
文献阅读 热心肠 SemanticScholar Geenmedical
扩增子分析:图表解读 分析流程 统计绘图
16S功能预测 PICRUSt FAPROTAX Bugbase Tax4Fun
在线工具:16S预测培养基 生信绘图
科研经验:云笔记 云协作 公众号
编程模板: Shell R Perl
生物科普:
来源:刘永鑫Adam
声明:本站部分文章及图片转载于互联网,内容版权归原作者所有,如本站任何资料有侵权请您尽早请联系jinwei@zod.com.cn进行处理,非常感谢!