基于GEMMA算法分析与细菌表型相关的基因型
- 1.介绍
-
- 1.1 介绍_简介
- 1.2 介绍_优点
-
- 1.2.1介绍_优点_排除了连锁不平衡的干扰3级标题
- 1.2.2介绍_优点_速度快
- 2.实际操作
-
- 2.1实际操作_分析流程概述
- 2.2实际操作_输入文件格式
-
- 2.2.1*.vcf文件
- 2.2.2*.bed文件
- 2.2.3*.fam文件
- 2.2.4*.bim文件
- 2.2.5id.txt文件
- 2.3实际操作_安装软件
- 2.3实际操作_流程
-
- 2.4.1实际操作_流程_基础文件夹配置
- 2.4.2实际操作_流程_质控和比对
- 2.4.3实际操作_流程_合并并形成*.vcf文件[6]
- 2.4.4实际操作_流程_转换格式
- 2.4.5实际操作_流程_单个基因和表型的关联
- 2.4.6实际操作_流程_基因和多个表型的关联
- 3.结果分析
-
- 3.1结果分析_文件
- 3.2结果分析_整合多个结果文件
- 3.3结果分析_绘图
- 4.参考资料
备注:本文同时发布在公众号“基因的生物信息学分析”,链接在这里
1.介绍
1.1 介绍_简介
GEMMA全称为:Genome-wide Efficient Mixed Model Association algorithm,即基于全基因组混合模型关联算法的工具[1]。GEMMA是一款基于混合线性模型的GWAS分析软件。可精确和快速地进行单SNP的GWAS、多SNP的GWAS和多性状的GWAS分析。
1.2 介绍_优点
1.2.1介绍_优点_排除了连锁不平衡的干扰3级标题
人类和细菌群落中本身的SNP差异造成了突变的连锁不平衡(即基因组本身序列有差异),造成了组内差异大于组间差异,进而导致通过GWAS分析找到的显著表达的基因只是由不同群体基因组的差异引起的,产生了大量的假阳性(图1)[2]。
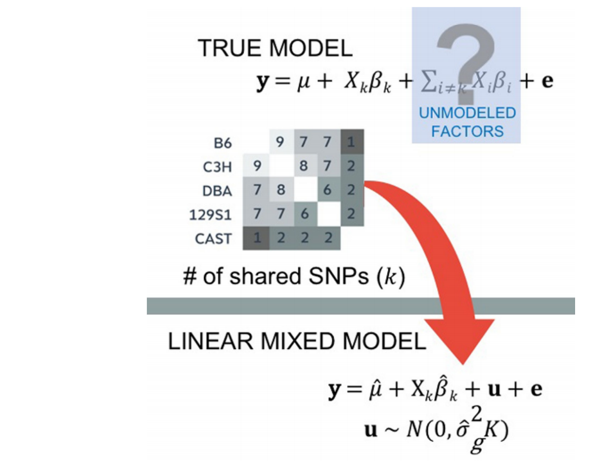
 图2.计算相似性矩阵并结合多元线性回归模型
图2.计算相似性矩阵并结合多元线性回归模型
1.2.2介绍_优点_速度快
使用固定的处理器配置对数据进行分析在比较中,GEMMA在精确算法中速度最快,在近似算法中也仅仅慢于GRAMMAR;GEMMA准确度和EMMA更接近(图3)[3]。但图3中只有一部分算法,新的算法如phylogenetic convergence test还在不断的被开发出来。
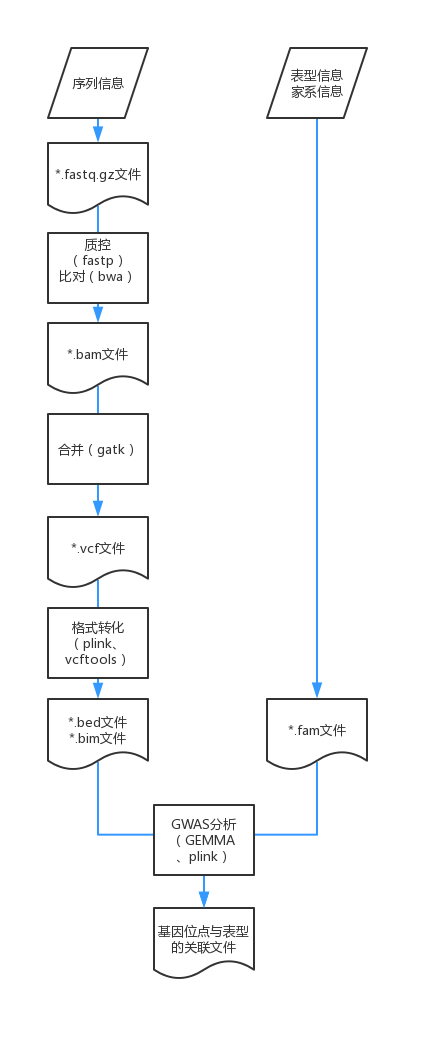
图4.流程
2.2实际操作_输入文件格式
GEMMA支持BIMBAM格式和plink二进制格式,主要包括以.bed、.fam、.bim。这3种文件需要由标准格式的.vcf文件转化而来。限于篇幅,只选取部分文件讲解格式。
2.2.1*.vcf文件
vcf(Variant Call Format)文件是存储变异位点的标准格式,可以用来表示单核苷酸多态性、插入缺失、结构变异、拷贝数变异等。因为vcf格式很复杂,本文中未提及的请查看https://samtools.github.io/hts-specs/或http://www.cog-genomics.org/plink/2.0/input#vcf。
前几行以“#”开头的行的含义:
| 简称 | 含义 |
|---|---|
| ##fileformat | VCF格式版本号 |
| ##FILTER | 显示这个文件已经进行了过滤 |
| ##contig | 参考基因组contig信息 |
| ##INFO | INFO列中各简写的含义。ID、Number、Type、Description主要有几个tag标记:AD、DP、GQ、GT、PL |
不以“#”开头的信息列的含义:
| 简称 | 含义 |
|---|---|
| CHROM | 表示变异位点位于哪个染色体 |
| POS | 变异位点相对于参考基因组所在的位置,若为删除或位移第一个碱基所在的位置。 |
| ID | 变异位点名称(对应dbSNP数据库中的ID;若没有,则默认用) |
| ALT | 该位点突变后的碱基类型类型,若有多个,则用逗号分隔。 |
| REF | 该位点参考基因组的碱基类型。 |
| QUAL | 可以理解为所call出来的变异位点的质量值Q。Q=-10lgP,P表示这个位点发生错误的概率。因此,当Q=20时,错误率P=0.01。 |
| FILTER | 针对质量值等变量过滤之后,在FILTER一栏都会留下过滤记录,通过过滤的位点FILTER一栏显示“PASS”;没有通过过滤的位点的FILTER一栏为其它信息。若FILTER一栏为“.”,说明没有进行过滤。 |
| INFO | 有关该位点的额外信息 |
| FORMAT | 变异位点格式,其中字母简写的含义在文件中以“#”开头的行中 |
| SMAPLE | 使用的样本名称,由bam文件中@RG的SM标签决定。对应的数据必定有GT(genotype,样本基因型)信息。 |
| GT:AD:DP | GT信息中,两个数字之间以斜线分隔则表示二倍体样本位于两条染色体上的基因型。0表示该位点的与参考基因组相同,1表示该位点存在一个突变,2表示改位点存在第2个突变。AD(Allele Depth)是样本中每一种allel的reads覆盖度,在二倍体中是用逗号分隔的两个数,前面对应ref,后面对应variant;DP(Depth)是样本中该位点覆盖度[4]。 |
示例:
##fileformat=VCFv4.2
##ALT=
##FILTER=
#CHROM POS ID REF ALT QUAL FILTER INFO FORMAT 081
NC_000962.3 1977 . A G 18395.03 . AC=2;AF=1.00;AN=2;DP=470;ExcessHet=3.0103;FS=0.000;MLEAC=2;MLEAF=1.00;MQ=60.00;QD=25.36;SOR=0.791 GT:AD:DP:GQ:PL 1/1:0,470:470:99:18409,1410,0
NC_000962.3 4013 . T C 16093.03 . AC=2;AF=1.00;AN=2;DP=400;ExcessHet=3.0103;FS=0.000;MLEAC=2;MLEAF=1.00;MQ=60.00;QD=28.73;SOR=0.776 GT:AD:DP:GQ:PL 1/1:0,400:400:99:16107,1202,0
2.2.2*.bed文件
包含基因型信息,以二进制格式存储(因为格式问题难以展示示例)。
2.2.3*.fam文件
包含表型信息,由六列组成的文本文件,从左到右分别为:
| 简称 | 含义 |
|---|---|
| 家庭号 | 没有则填“0” |
| 个体号 | 即样本号,不能为“0” |
| 父亲号 | 没有则填“0” |
| 母亲号 | 没有则填“0” |
| 性别 | “1”=男 , “2”=女, “0”=未知 |
| 表型 | 0=对照组, 2 = 实验组, -9/0/非数字 = 缺失数据) |
示例:
081 081 0 0 0 1
0 278 0 0 0 1
0 279 0 0 0 1
0 283 0 0 0 0
0 289 0 0 0 1
0 291 0 0 0 1
0 299 0 0 0 1
2.2.4*.bim文件
存储基因型和位置等信息,一个包括6列的文件。
| 简称 | 含义 |
|---|---|
| 染色体号 | 染色体的类型或名字,可以为“X”,“XY”、“Y”、染色体的名字等,若无则填“0” |
| 突变编号 | 突变的标记,一般为.以摩或厘摩为单位的位置 若无则填“0” |
| 碱基对的系数 | Base-pair coordinate (1-based; limited to 231-2) |
| 次等位基因 | .bed文件中对应的次要等位基因(突变基因型) |
| 主要等位基因 | .bed文件中对应的主要等位基因 |
示例:
0 . 0 21 CAGT AGGC
0 . 0 29 C A
0 . 0 30 G C
0 . 0 42 T C
0 . 0 43 G A
2.2.5id.txt文件
为了批量处理文件,将样本id分别作为一行,写入文件。
示例(id.txt):
081
082
2.3实际操作_安装软件
| 安装项目 | linux有关命令 |
|---|---|
| 安装gatk | wget https://github.com/broadinstitute/gatk/releases/download/4.1.4.0/gatk-4.1.4.0.zip unzip gatk-4.1.4.0.zip ; cd gatk-4.1.4.0 conda install git-lfs -y git lfs install git lfs pull –include src/main/resources/large echo ‘export PATH=”/data/home/lizhiyuan/biotools/gatk:$PATH”’ >> ~/.bashrc |
| 安装bwa | wget https://jaist.dl.sourceforge.net/project/bio-bwa/bwa 0.7.17.tar.bz2 tar -xjf .tar.bz2 cd bwa- make |
| 安装plink | conda install plink -y |
| 安装GEMMA | conda install gemma -y |
2.3实际操作_流程
2.4.1实际操作_流程_基础文件夹配置
创建每一步需要的文件夹,并把数据文件和参考基因组文件分别拷贝进00_data文件夹,结构如下图

你可以在这里找到更多关于bwa软件使用的信息5.
2.4.3实际操作_流程_合并并形成*.vcf文件[6]
2.4.4实际操作_流程_转换格式
2.4.5实际操作_流程_单个基因和表型的关联
2.4.6实际操作_流程_基因和多个表型的关联
3.结果分析
3.1结果分析_文件
产生的结果文件merge_plink_assoc.assoc.txt中,从左到右分别为:
| 简称 | 含义 |
|---|---|
| chr | 染色体 |
| ps | 突变位点的位置 |
| n_mis | 缺失数据的位点数,如果这个数过大,说明文件格式可能错误 |
| n_obs | 有数据的位点数,如果这个数过小,说明文件格式可能错误 |
| allele1 | 次要等位基因(突变位点的基因型) |
| allele0 | 主要等位基因(参考基因组的基因型) |
| af | 次要等位基因频率 |
| beta | 基因对表型的影响大小,即线性回归模型的斜率 |
| se | beta的标准差 |
| p_wald | 使用wald检验(或其它方法)计算出的显著性大小。越小结果越显著。 |
示例:
chr ps n_mis n_obs allele1 allele0 af beta se p_wald
0 4187485 5 4271 TG CG 0 -4.88E-01 7.64E-02 1.86E-10
0 3829770 12 4264 T C 0 -4.89E-01 7.64E-02 1.82E-10
3.2结果分析_整合多个结果文件
可以使用python中的pandas[7]包高效整合不同*assoc.txt文件
合成的文件格式如下:

3.3结果分析_绘图
根据得到的P值,可以使用R包qqman [8] 和ggplot2[9]分别绘制QQ图曼哈顿图,QQ图中点和线越靠近结果越好,曼哈顿图中位点的数据点位置越高越显著,有关二图图注含义的解释可以查看参考资料[10]。
library(qqman)library(ggplot2)setwd("08_results/01_assoc")gwasResults1 来源:寰宇尽头遥望璀璨的天眼
声明:本站部分文章及图片转载于互联网,内容版权归原作者所有,如本站任何资料有侵权请您尽早请联系jinwei@zod.com.cn进行处理,非常感谢!