目录
- 前言
- 文章内容
- 一、文章介绍
-
- 1.目的
- 2.背景
- 二、十个建模技巧
-
- 1.模板选择
- 2.阅读发表的相应模板三维结构的文章
- 3.去除不必要的元素、溶剂、配体和离子
- 4.优化3D结构,如果氢丢失要补回氢
- 5.留意比对过程的gaps
- 6. 进行loop建模时要考虑loop大小以及残基组成
- 7.在进行计算分析前,进行结构最小化是必不可少的
- 8.使用多种策略进行三维结构的验证和评估
- 9.考虑氨基酸质子化作用
- 10.了解拓扑结构
- 建模非蛋白质分子
- 案例研究
- 最后说两句
前言
博主刚把脚踏进生物物理和生物信息这领域,本身没有坚实的生物基础,也没有专门去学计算机,所以在这领域中撞墙碰壁,苟延残喘。如今想要开始通过写博客的方式,在平台上向各位大佬各位学者学习。我觉得知识是要拿出来分享,不仅帮助了他人,也充实着自己。
我打算隔一段时间写一篇博客关于生信中同源建模相关(或是其他)的文献,我也是第一次解读文章编写博客,会有很多不足的地方,各位看官若能够指出我哪些做得不够的地方,或是指出文献解读方面不足之处,那就再好不过了,这对我的帮助很大,在这先谢谢大家了哈!
今天要讲内容的是一篇入门蛋白质同源建模的文献——《Ten quick tips for homology modeling of high-resolution protein 3D structures》。这篇文献偏介绍性,例如建模的基本流程,在各步骤需要留意的点或处理技巧,以及介绍在建模过程中哪些地方可以使用哪些软件。整篇来说,概念性较强,对初学同源建模的同学还是有帮助的。
下面要讲的这篇文章是《Ten quick tips for homology modeling of high-resolution protein 3D structures》,链接[https://doi.org/10.1371/journal.pcbi.1007449]
文章内容
接下来将要对文献进行简要讲解,我会挑出每个Tip中我觉得有帮助的点,并且尝试去多讲一些。
一、文章介绍
1.目的
文章作者想给初学建模的人通过介绍的系统实践的方法来建模出高分辨率的蛋白质3D结构。建模者要学会能够访问和使用原子坐标来构建同源模型。另外想要提供一个原理就是在建模的基础上制作一个简单的原子坐标列表,用于符合物理原理的计算分析。后面还有一部分内容是关于建模非蛋白分子和同源建模的例子实践。
2.背景
根据蛋白质能量景观漏斗假说:蛋白质native结构应该在漏斗低端具有最低的自由能,即全局能量最小。如今就有很多计算策略通过查找势能景观来确定蛋白质native构象,这些想法被分为两种算法:一种是确定性的(deterministic),一种是启发式的(heuristic)。区别是搜索构象空间的coverage的不同。
确定性方法扫描整个或者大部分构象空间,基于先验知识排除掉子空间,(举例:同源建模允许修改同源结构来预测蛋白质3D结构,这样就可以消除掉大量的构象。)注意:大师兄说现如今好像没有可以扫描搜索整个构象空间的方法,即使是同源建模。其实我觉得构象空间这个概念很抽象,感觉言语上说不清。
启发式方法只搜索构象空间一小部分,但有一组代表性的构象,例如MD应用能量函数研究力,求解运动方程,并预测原子轨迹。尽管c空间覆盖有限,MD提供了关于折叠和展开路径的信息。这些方法常用于工业酶以及药物制造中。
同源建模工作流程分为以下几个步骤,开始于选择最佳模板3D结构,第一次的序列比对通常使用BLOcks替换矩阵执行。第二次序列比对(也称为比对校正)用于构建骨干三维结构。然后对无模板区域或者相似性比较低的区域进行loop建模。最高精度可达12~13个残基。接着是侧链重建,通过依赖主链的旋转体库进行构象搜索。接下来应该通过各种质量评估工具对结构进行改进和验证。注意:序号代表的是在哪几个Tip有提及到。
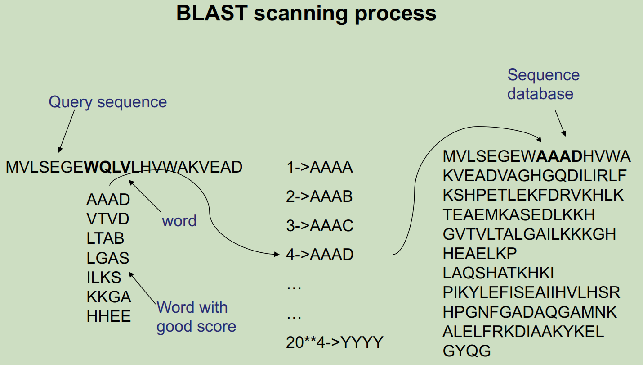

PSI-BLAST:跟BLAST一样需要一段高打分片段(HSP),以此片段开始往两方向扩展匹配。
Blast和PSI-Blast的区别:
1.PSI-Blas用2个HSP而blast用1 个HSP。
2。PSI-Blast中动态规划扩展的HSP允许gap。
3.PSI-BLAST是Profile-sequence对齐,而不是sequence-sequence对齐。
4.迭代。
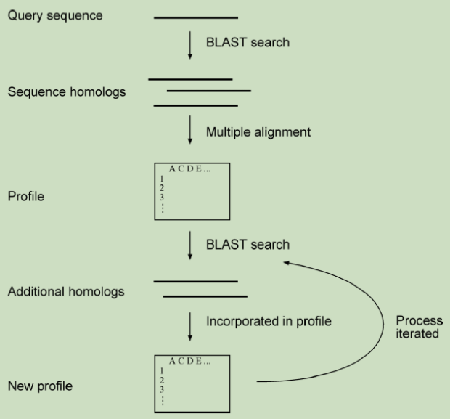
Profile文件(下文称参数文件)是对多序列比对一致序列的描述(如下图)。它使用一个位置特定的评分系统来捕获关于多重对齐中各个位置的保守程度的信息。这使得它比使用位置无关评分的成对方法(如BLAST或FastA使用的方法)更敏感、更具体。
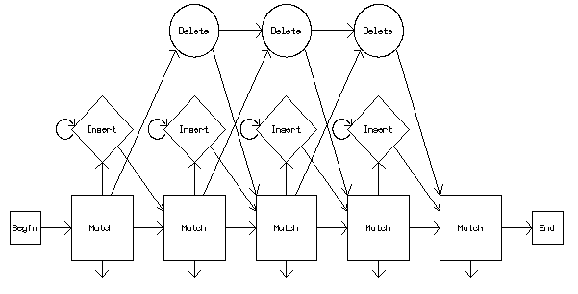
虽然Profile HMM需要更多的计算,但因为可以检测对齐中的空隙,它们对于低同源性的情况更有用。一般来说,建议使用相似性低于35%的Profile HMMs和相似性高于35%的PSSM比对以及序列序列比对。同源性建模中的比对gap或“移位”非常关键,序列比对出错会对主链产生影响,进而在侧链放大,确定了gap则可以准确地重新进行建模。注:这里介绍的内容都比较浅显,若需要使用某一方面方法则需要自己进一步学习。
6. 进行loop建模时要考虑loop大小以及残基组成
一般来说,插入和删除很少出现在a螺旋和β折叠片上,经常是出现在多变且不保守的loop结构上。Loop结构对功能特异性很重要。如何进行loop建模取决于循环的大小和氨基酸组成,它们直接影响构象空间的扫描。在组成方面,最关键的残基变化通常是从任何一个残基到脯氨酸,从甘氨酸到任何一个残基。在这两种情况下,两个残基必须符合更严格的骨架二面角范围。
常用的几种loop建模方法以及可使用的软件:
1.Ab intio:该方法使用穷举搜索,通过使用基于知识的评分函数的优化算法识别最小能量构象。算法精度在不同decoy上面进行测试,枚举法就是其中一种使用的是短寡肽构象虚拟数据库。从头开始的方法最大的一个挑战就是如果loop增加一个新氨基酸,其构象空间指数爆增。MODELLER和Rosett里包含有loop从头建模的方法。
2.Knowledge-based:这种方法筛选x射线晶体学数据库,以找到给定循环序列的同源构象。Swiss-model里面ProMod3、FREAD、DaReUS-Loop(基于数据的方法,使用远程或不相关的结构进行循环建模)都有很好的基于知识的loop建模方法。
3.组合策略:即组合优化从头建模算法和基于知识的统计势,以达到更高的精度。对于长循环(十个残基以上),建议采用不同的初始方法进行采样,然后进行总体评分。
7.在进行计算分析前,进行结构最小化是必不可少的
为了能够将物理定律应用在3D建模计算上,研究员使用力场来保证准确的原子质量、电荷以及原子键。所谓力场就是一套描述键长、键角、二面角、非正常平面、静电和范德华力的标准参数和方程。从数学上讲,三维结构最小化的过程是通过多变量算法对势能面进行几何优化来实现收敛的。最抖下降法(SD)和共轭梯度(CG)法都是基于梯度的迭代方法,使用算法中目标函数的一阶导数值(斜率)来寻找最小值。
SD方法是最简单的算法,但它的收敛性相对较差,经常在最小值附近循环,很少能达到最小值。它主要用于在MD模拟之前减少原子冲突,或者作为更复杂的最小化策略的第一步。CG方法通过对梯度和前一次迭代的方向进行加权平均(从而补偿缺失的信息)来存储之前的梯度,并且具有比SD方法更好的收敛性。牛顿-拉夫森(NR)方法在其算法中使用一阶导数和二阶导数,并给出了与CG方法比较的结果。
一个好的最小化protocal是结合两个或两个以上的算法:例如,首先使用SD,然后使用CG或NR。检查势能面是否收敛有四个标准:1.力要为零。2.力的均方根为零。3.下一步优化的位移小于截距值或者4.下一步优化的位移均方根小于截距值。
几乎所有的MD程序、图形界面可视化软件和分子建模程序都有整合最小化集成包。例如使用MD程序的AMBER和GROMACS输入文件指定最小化参数:最小化方法,迭代次数,截距标准及其值。图形可视化软件例如Chimera,Pymol和VMD,最小化参数在窗口和插件设置中定义。
8.使用多种策略进行三维结构的验证和评估
建模软件会根据各种评分方法对产生大量的3D结构进行评估排名。因为不同评估方法从不同角度对模型进行评估,所以结合多种方法对模型评估更加有利。评估方法可分为四类:
1.基于物理的方法:大多数这种方法是基于力场和优化立体化学的计算。MolProbity对蛋白质x射线晶体结构进行评估,从全局和局部角度验证其质量。这种方法可以检测骨干异常值(Ramachandran和Cβ偏差)、侧链异常值(旋转异构体偏差)还有不正确的全原子接触(原子冲突)。骨干异常值的出错会被放大,Ramachandran异常值描述二面角Ψ和Φ的偏差,也就是蛋白质的二级结构。Cβ异常值表示Cβ原子位置的偏差。另外类似的程序还有WHAT IF和PROCHECK。
2.基于知识的方法:这是依赖数据库的验证方法,使用的分数表示在数据库中所有已知的实验3D结构中统计获得的能量。例如SWISS-MODEL使用QMEAN(Qualitative Model Energy ANalysis)对Cβ相互作用能、全原子成对能、扭转角能和溶剂化能进行打分。MODELLER建模软件使用两种基于知识的方法进行打分,一种是DOPE(Discrete Optimized Protein Energy 离散优化蛋白质能量),另外一种是PROSAII,还有一种基于物理的评估方法(PROCHECK)。质量评估(QA)-重新组合服务器允许用户在共识的基础上建模蛋白质三维结构,在广泛的输入蛋白质三维模型中识别高度保守的区域。通常会采用几个方法对单个模型或者聚类得到的几个模型进行评分。
3.基于机器学习的方法:利用支持向量机(SVM)回归方法,建立对同源模型预测误差的复合评分。支持向量机是一种监督学习算法,它从训练数据集导出特征,并在单独的数据集上进行测试,这对于回归、分类或聚类都是有用的。
4.基于实验的方法:实验验证(保留分辨率)是理论模型的最终测试。评价三维同源结构最简单的方法是均方根偏差(RMSD),它给出了两个三维结构中所有原子之间距离的平均值。由于结构域之间loop的最小扰动会导致误导的高RMSD,因此首先将蛋白质分割成片段更好地应用该方法。SphereGrinder在三维模型中基于选定原子的局部模体中进行基于RMSD的测试。根据不同的质量水平,由用户定义的半径球体来表示本地化的图形。还有一种更系统、更准确的方法来考虑局部(较小)区域,并进行局部和全局结构叠加。 这种方法称为全局距离(GDT),CASP实验中应用这种方法评估模型。
9.考虑氨基酸质子化作用
在化学中,质子化(或水化)是将一个质子(或水合氢,或氢阳离子)(H+)加到一个原子、分子或离子上,形成共轭酸。通常侧链的质子化在蛋白质中的大部分氢供体和/或氢受体和静电相互作用中起着作用,并会影响计算预测的准确性。质子化受周围环境的影响,包括pH、温度和相邻残基。质子化以及去质子化将导致侧链上产生不同的静电电荷,这将强烈影响力场参数,导致相互作用。H++服务器通过对连续体静电学和分子力学的研究估计pK常数来预测质子化。质子化对于预测药物和底物在结合位点内的位置分子对接的准确性和传统的MD研究具有重要意义。研究质子化的方法有:杂化量子力学和分子机制(QM/MM)以及经验价键(EVB)。在量子力学中,原子分为量子力学系统(模拟电子及其相互作用)和分子力学系统。EVB不需要模拟电子,只应用原子的部分电荷来计算价键共振形式的能量。
10.了解拓扑结构
FF包括标准参数(描述拓扑)和与三维结构相关的计算所涉及的方程。应用力场估计原子和键上的力称为分子力学。最初的力场计算为了节省计算成本,并没有明确地使用氢,而那些明确使用所有原子的被称为全原子力场。分子力场的参数,也称为拓扑,是从实验和计算化学方法中导出的。对于每个力场,根据不同应用程序,可以使用不同的拓扑集。
有些立场缺乏DNA、脂类、碳水化合物或离子的拓扑结构,以前的FFS使用不同的原子类型来描述同一元素的各种拓扑(键长;角度)。一种现代方法通过直接化学感知的引入,对FF新定义。与传统的原子类型相反,通过基于分子中的子结构来分配拓扑。
对于MD,您越早学习如何读取和编辑拓扑文件,您就可以越快地在该领域前进,并模拟复杂的分子。
翻译后的修饰(PTMs):侧链PTM在过去一直被忽略,直到Rosetta程序开始在建模中加入非标准氨基酸。SIDEpro server、PyTMs program均是预测翻译后修饰的程序。识别翻译后修饰的残基也有助于验证和改进循环建模。
非共价配体:大多数MD软件可以根据一般力场参数生成配体拓扑结构。例如AMBER的Antechamber和CHARMM。还有其他专门服务器和数据库准备就绪拓扑文件,例如场拓扑生成器的自动化(ATB)和PRODRG。拓扑文件的生成,也称为参数化,是通过量子力学计算完成的。 这些计算包括以下几点:(1)先是在低理论水平上优化配体( 半经验分子轨道水平)。
建模非蛋白质分子
相比蛋白质、核酸、脂类和碳水化合物,过渡金属离子、反应底物和小药物或配体的拓扑结构在力场中的情况则更为复杂。这里介绍非标准残基如何处理。
水分子模型:水通过氢键在蛋白质功能中起着重要的作用。固定的水分子可以在配体结合位点或两种相互作用的蛋白质之间的界面上找到,并能影响分子对接的准确性。经常用的MD模型是刚性3位点,例如TIP3P模型,可以根据FF修改拓扑。水模型之间的微小差异取决于范德瓦尔斯和静电分量。对于MD模拟,使用与FF兼容的水模型是很重要的。
翻译后的修饰(PTMs):侧链PTM在过去一直被忽略,直到Rosetta程序开始在建模中加入非标准氨基酸。SIDEpro server、PyTMs program均是预测翻译后修饰的程序。识别翻译后修饰的残基也有助于验证和改进循环建模。
非共价配体:大多数MD软件可以根据一般力场参数生成配体拓扑结构。例如AMBER的Antechamber和CHARMM。还有其他专门服务器和数据库准备就绪拓扑文件,例如场拓扑生成器的自动化(ATB)和PRODRG。拓扑文件的生成,也称为参数化,是通过量子力学计算完成的。 这些计算包括以下几点:(1)先是在低理论水平上优化配体( 半经验分子轨道水平)。(2)在更深层次的理论水平(例如,从头计算水平,如Hartree-Fock分子轨道或密度泛函理论水平)计算静电势,以获得更高的精度。最终验证可以通过将结果与配体的NMR数据进行比较来实现。
反应底物和共价配体:研究反应底物和共价配体反应的黄金标准是混合QM/MM方法,它可以更好地描述化学键的断裂和形成,特别是在酶和底物反应的研究。经典MD不能预测共价键的形成或断裂。
金属离子:除了QM/MM方法外,还可以在经典的MD模拟中表示金属离子,可以只在非键合态(仅包括静电和范德瓦尔斯拓扑)、键合态(保持键、角度),二面体、静电和范德瓦尔斯拓扑),或虚拟原子状态,通常构成许多用户定义的交互位置。
案例研究
使用SWISSMODEL服务器从单个模板中建立一个蛋白质结构域的同源性模型,并在UCSF Chimera程序中对其进行优化,并且使用MolProbity和QMEANSE的质量信息评估模型。目标序列是ALK间变性淋巴瘤激酶的一个结构域MAM1,Uniprot ID:Q9UM73。这里还需要从激酶序列中提取截取对应结构域的序列。最开始将目标序列提交到SwissModel服务器上进行模板搜索,找到四条模板序列,这里比较有意思的作者选择的是coverage高的序列而不是序列相似性高的序列。2c9a.1.A(1-163/0.89),,2v5y.1.A (2-156/0.86),,5l73.1.A (1-162/0.88),,5l73.1.B (1-162/0.88)。最后面数值指的是coverage。
注意:这里coverage与之前的构象空间的coverage又不相同,具体意思没有理解清楚。思考:为何是选取coverage高的序列而不是选取序列相似性高的序列。
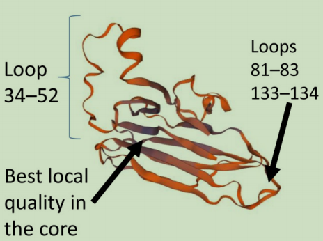
再将其进行结构评估,可以得到异常值的Ramachandran点图,MolProbity分析以及QMEAN点图。

Unfortunately, all loop models were stretched-loop shaped. In this unique situation where we speculate the actual model of the loop, the best decision would be to use this ab-initio model as a second template for homology modeling.根据作者意思,所有的loop模型都是拉伸环路形状,作者推测loop的实际模型特殊情况下,最好是使用这个从头算模型作为同源建模的第二个模板。注:其实这里并不很理解,单单从这个作为第二模板构建同源模型来说,如何实现的可行吗者并没有给出使用这个从头算模型作为同源建模的第二个模板构建模型的步骤算法,这里估计只是推测。
然后使用Chimera根据QMEAN得分低的区域进行最小化。
Favorites>Sequence ,Tools>Structure Editing>Minimize Structure using 3,000 steps SD then 1,000 steps CG
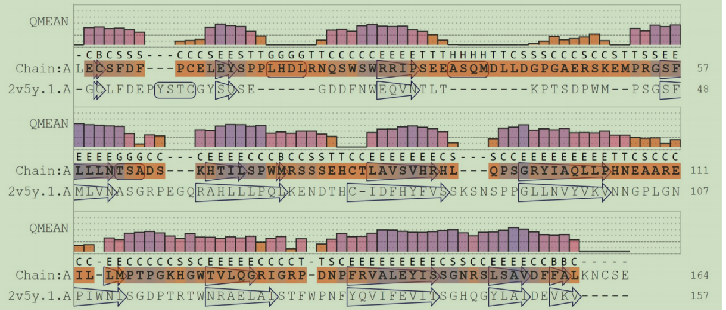
再提交到SwissModel服务器中,重新检查需要改进的剩余残留物。将对应Ramachandran异常值的几个氨基酸(Pro15, Asp19, Pro31, Ala 46, and Pro53)进行旋转操作(Tools>Structure Editing>Rotamers)。对于氨基酸的处理需要注意。最后再将整个蛋白质模型进行最陡梯度下降法跑一遍。最后得到的是QMEAN得分为-3.20,比之前的降低些。但是Molprobity和clash分数从最初的2.45,10.93下降到1.81,1.62。这里师兄建议将最后得到的模型与PDB库的原始蛋白质结构进行比较,才知道是否是正确的。但是这个目标序列没有实验结构,所以无法证明上述步骤建出的模型是好是坏。
最后说两句
从一个初学者角度来说,文章确实可以为读者提供很多建模思路,解决方法,处理软件以及需要注意地方,有些地方也是让我理解不了作者的想法,归根到底是自己知识面窄,总之读完后还是有很多的疑难困惑,在文中简单表明了几个问题,如果说您在读的时候也有问题想法或是解决办法,可私信可留言,请多多指教。若是有幸,希望大家能够留言提出问题,提出想法,提出意见,解答疑惑。这对大家会有帮助的,感谢感谢!
来源:阿泽
声明:本站部分文章及图片转载于互联网,内容版权归原作者所有,如本站任何资料有侵权请您尽早请联系jinwei@zod.com.cn进行处理,非常感谢!