关注泰科科技 做模拟不迷路


摘 要
为了探究重晶石的可浮性规律,采用Materials Studio软件CASTEP模块对重晶石的原胞及解理面(001)面进行了表面基因特性研究。首先对重晶石原胞进行了结构优化,根据与重晶石实际晶胞参数比对的方式确定了优化参数中的密度泛函为GGA-WC,k点取样为4×3×4,截断动能为530eV。基于优化后的重晶石原胞创建(001)面,然后计算(001)表面电子态密度、Mulliken 电荷布居、键布居及表面能等表面基本化学特性。计算结果表明:重晶石原胞中Ba—O键比S—O键的键长更长,因此断定Ba—O键键能更小,断裂的概率更大;通过对(001)面上各原子的态密度与Mulliken 电荷布居分析,可判定Ba2+、—O-为(001)面上主要的具有化学活性的位点。重晶石晶胞表面基因特性研究可为重晶石浮选过程中的表面活性位点的判定提供基础,进而可以对矿物可浮性与浮选药剂的作用机理研究提供借鉴与参考作用。
引 言
重晶石是重要的非金属矿物原料,在工业中具有广泛的用途。我国已探明的重晶石储量80% 以上是以伴生矿形式存在,贫矿较多,对其中嵌布粒度较细的重晶石矿及含重晶石的重选尾矿,常采用正浮选的方式进行分选回收。浮选中的吸附过程与矿物的表面性质紧密相关,采用基于密度泛函理论的量子化学计算方法对矿物晶体表面化学特性进行表征已经是科学工作者的常用手段;将理论计算与实际试验相结合,可以为矿物浮选过程中与药剂分子的作用机理研究提供很好的指导与参考作用。
重晶石的主要化学成分是硫酸钡(BaSO4),其晶格通常为斜方晶系,晶体的(001)面是重晶石最常见解理面。本研究采用基于密度泛函理论的CASTEP模块,模拟了重晶石的(001)表面,并通过计算该表面的表面能、态密度、Mulliken电荷布居、键布居来表征重晶石的表面基因特性,以判定(001)面上主要化学活性位点,为重晶石浮选过程中与浮选药剂的作用机理研究提供参考。
模拟与计算方法
1. 重晶石晶胞模拟方法
采用Materials Studio软件2017R2版本的CASTEP模块对重晶石晶胞结构进行优化,并将模拟结果和实验参数进行比较,验证优化后晶体结构在晶胞参数与原子占位上的准确性和可靠性。矿物原胞内原子Mulliken键布居的计算则基于几何优化后的模型上进行;在该计算中,价电子和核的相互作用都采用OTFG-超软赝势来描述;几何优化原子间作用力收敛阀值为0.05eV/nm,原子位移收敛阀值为0.0002 nm,体系总能量的变化收敛阀值为20μeV/atom,原子间的内应力收敛阀值为0.1 GPa;并且所有的计算都在倒易空间中进行。
2. 交换相关泛函的选择方法
为确定CASTEP模块优化重晶石晶胞结构的最佳参数,首先对计算中所使用的DFT方法、布里渊区k 点采样与截断动能进行考察,k点取样2×3×2、截断动能571.4eV时,不同DFT 方法优化重晶石晶胞的模拟结果对比见表1。

由表1 可知,采用GGA-WC方法计算获得的晶格参数与实际检测值相差最小,偏差小于1%。因此选用GGA-WC作为计算中使用的DFT方法。
3. k点取样与截断动能参数的选择方法
进一步对重晶石晶胞结构优化计算的k 点取样与截断动能参数设置进行考察。分别改变k 点取样和截断动能参数设置,计算重晶石晶胞体系能量与晶胞参数,如图1、图2所示。由图1 与图2计算结果可以看出,在k 点取样设置在4×3×4以上,截断动能在530eV 以上时,所得到的晶胞结构与体系能量基本稳定,因此综合考虑模拟可靠性与计算效率,模拟计算的k 点取样选择4×3×4,截断动能选用530eV。
4. 表面能的计算方法
表面能是指在外力作用下沿某一晶面方向使晶体解理断裂成2个独立表面所需能量,其大小取决于表面原子间的相互作用,与表面原子的几何结构密切相关。表面能越小,说明表面的稳定性越高。表面能计算公式如式(1)所示。
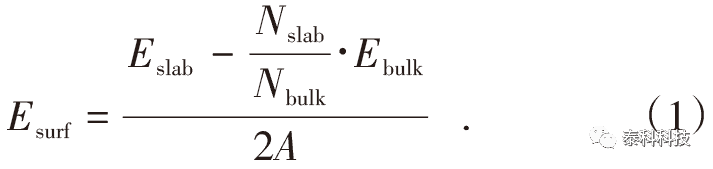
式中,Esurf为表面能,J/m2;Eslab和Ebulk分别为表面模型和原胞的总能量,J;Nslab和Nbulk分别为表面模型与原胞模型的总原子数,A为表面模型沿轴方向的面积,m2;2表示表面模型沿轴方向有上下2个表面。

试验结果及讨论
1. 重晶石晶胞的优化与表征
采用CASTEP模块,在密度泛函选用GGA-WC、k点取样4×3×4、截断动能530eV条件下,对重晶石晶胞进行结构模拟。优化后的晶胞结构如图3 所示。

优化后的晶胞模型的模拟XRD与实验测试重晶石纯矿物XRD图谱对比,结果如图4所示。
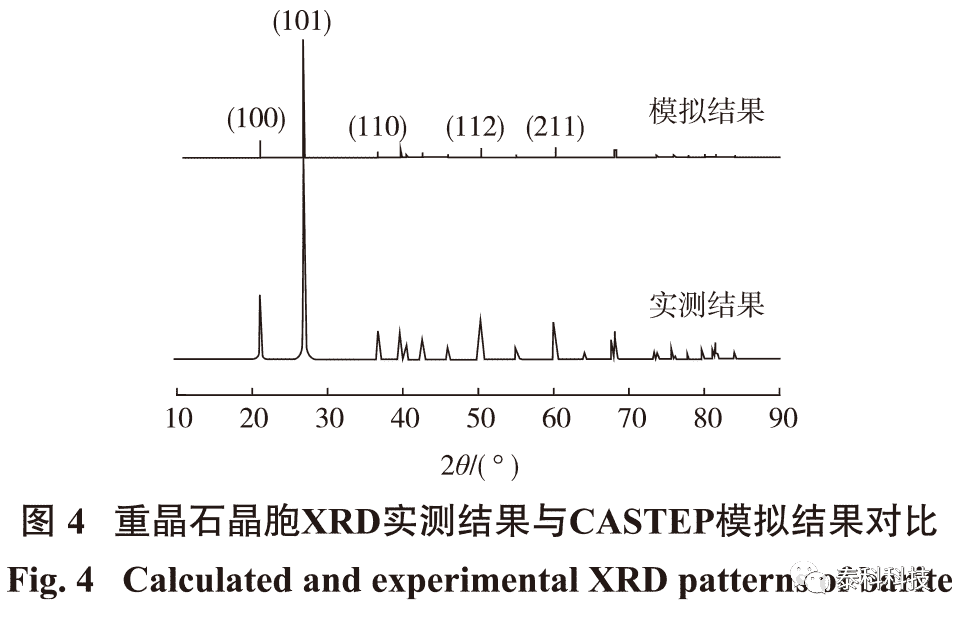
从图4 可知,模拟所得XRD图谱与纯矿物检测图谱吻合较好,模拟结果贴合实际矿物晶胞,表明试验参数选择有足够的可靠性。重晶石原胞不同种类键的Mulliken布居值如表2所示。

从表2 可以看出:S―O键布居值变化范围为0.540~0.580,键长变化范围为0.147 7~0.1503nm,为共价键;Ba―O键布居值变化范围为0.050~0.070,键长变化范围为0.2798~0.2910nm,为典型的离子键。可以推断,Ba―O键键能更低,断裂概率更大;S―O键键能较大,断裂概率较低。因此重晶石表面S出现概率更低,Ba和O出现的概率更高,在重晶石矿物表面更应该关注Ba和O的物理化学特性。
2. 表面模型的构建及其表面能的计算
在优化晶胞基础上,构建重晶石(001)面表面模型。根据不同的Top位点,重晶石(001)面有2种不同的切割方式。如表3所示Top位点为0.342(比例值,无量纲)时,所建立的表面模型的表面能能量远小于Top位点为0.658的表面能,因而在自然环境中更可能稳定存在,故选取Top位点0.342作为表面模型的切割位点。

表4 为真空层厚为2nm时,不同切割层数重晶石(001)面表面能,表5为切割层数为3层时,不同真空层厚度重晶石(001)面表面能。


从表4、表5可以看出:能量态最低时,切割层数为3层,真空层厚为1.8nm,因此选用3层与1.8nm作为切割层数与真空层厚度。然后在top位点为0.342、切割层数为3层、真空层厚度1.8nm的条件下创建重晶石表面模型,根据式(1),得到重晶石(001)表面能为0.1386 J/m2。
3. 重晶石(001)表面结构模拟与表面原子态密度
根据1.3节的表面模型的构建方法,在Top位点为0.342、切割层数为3层、真空层厚度为1.8nm的条件下,得到重晶石(001)表面模型,优化后的重晶石表面结构如图5所示。重晶石(001)表面原子如图6所示。
图6中重晶石(001)面表面第一层原子的态密度图.如图7所示。该表面原子的Mulliken 电荷布居结果如表6所示。
从图7可以看出:重晶石(001)面表面第一层原子的态密度在费米面附近主要是由表面氧原子的2p轨道电子密度贡献,而在费米面以上的导带底主要是由钡原子的d轨道和硫原子的p轨道电子密度贡献,表明在重晶石(001)表面上的氧原子有较高多电子活性,在参与化学反应时为电子供体。



根据以上重晶石晶体表面基因特性研究结果可以看出,低能面(001)的表面具有化学活性的是:①钡离子缺电子活性位点,②氧负离子的多电子活性位点。这些化学活性位点交错排列贯穿整个(001)表面。因此,在重晶石浮选体系中,凡是与Ba2+能够发生反应的阴离子,都是对重晶石矿物表面能够起到调节作用的物质,如CO32?(K?sp=2.6′10-9)、SO2-4 (K?sp=1.1′10-10)、PO3 -4(K?sp=3.4′10-23)等;凡是与—O?能够发生反应的物质,也是对重晶石矿物表面能够起到调节作用的物质,特别是—O-又是很好的配体,其可以与d区金属离子发生配合作用。总而言之,重晶石晶体表面基因特性是重晶石浮选过程中需要研究和考虑的最本质特性。
从表6 可以看出:—O-的Mulliken 电荷布居值为-0.92e或者-0.94e,为多电子位点,是电子供体;Ba离子的Mulliken 电荷布居值为1.43e 或者1.44e,为缺电子位点,S原子由于与—O-形成σsp3-p共价键被包围在四面体中心,不裸露在表面,因此不是重晶石的表面活性位点。

结 论
(1) 在重晶石原胞结构优化中采用GGA-WC、k点取样为4×3×4、截断动能为530eV 时,计算获得的晶格参数与实验检测参数相差最小,所得到的晶胞结构与体系能量最稳定。基于优化的重晶石原胞在top位点为0.342、切割层数为3层、真空层厚度1.8nm 的条件下创建(001)表面,低能面(001)的表面能为0.1386 J/m2。
(2) 重晶石原胞S―O键布居值变化范围为0.49~0.61,键长变化范围为0.1468~0.1523nm,为共价键。O―Ba键布居值变化范围为0.04~0.11,键长变化范围为0.1744~0.2919nm,为典型的离子键。可以推断,O―Ba键键能更低,O―S键键能较大。
(3) 重晶石的(001)面表面第一层原子的电子态密度在费米面附近主要是由表面氧原子的2p 轨道电子密度贡献,而在费米面以上的导带底主要是由钡原子的d 轨道和硫原子的p 轨道电子密度贡献,表明在重晶石(001)表面上的氧原子有较高多电子活性,在参与化学反应时为电子供体。
(4) 重晶石(001)表面化学活性位点为钡离子缺电子活性位点与氧负离子多电子活性位点。药剂分子可通过与两者结合来改变重晶石表面的亲水或亲油能力,进而影响矿物的可浮性。
文章详情:
10.19614/j.cnki.jsks.202006014

北京泰科涉及行业
材料研发
基于BIOVIA Materials Studio材料设计平台,提供涉及电池、航空航天、国防军工、建筑、涂料涂层等多领域材料研发软件及综合解决方案。
药物研发
针对药物设计、药物研发等提供基于Discovery Studio、COSMOLOGIC等软件的ADME、构象比对、溶剂筛选、结晶、成盐、共晶筛选、稳定性、溶解度pKa、分配系数等性质的模拟预测软件及方案。
化工设计
面向精细化工、新能源、石油化工等领域提供精馏萃取催化剂设计、热力学性质(溶解度、粘度等)、提纯表面处理吸附等性质模拟软件平台及解决方案。
数据挖掘
基于Pipeline Pi
来源:北京泰科博思科技有限公司
声明:本站部分文章及图片转载于互联网,内容版权归原作者所有,如本站任何资料有侵权请您尽早请联系jinwei@zod.com.cn进行处理,非常感谢!