关注北京泰科 做模拟不迷路



01
摘 要
利用量子力学从头算方法对硒化铍(BeSe)从分子、体块、单层(h-BeSe)到单壁曲折(n,0)和纳米管(SWBeSeNTs)的不同排列进行了广泛的计算研究。对于分子,作者使用高度相关的从头算方法进行了电子计算,这里是在多组态自洽场和多参考组态相互作用水平上,包括Davidson修正(MRCI+Q)和MOLPRO程序中实现的aug-cc-pV5Z基组,得到了精确的电子基态和激发态势能曲线,导出了一组振动光谱参数。
引 言
02
最近,低维材料已经成为科学界在新纳米技术投资方面的一个备受争议的问题。其中,石墨烯纳米片和碳纳米管因其特殊的电子结构和极低的带隙而备受关注。纳米材料的一个主要挑战是预测和优化它们的单一和二维纳米结构,如纳米线和纳米管。在这方面,对其结构和振动特性的探索已成为将其应用于纳米尺度器件的重点。本文采用多组态自洽场、包括Davidson修正(MRCI+Q)在内的多参考组态相互作用和带有B3LYP泛函的密度泛函理论等方法对第一性原理进行了研究。研究目标是不同形式的BeSe的结构和振动特性,从3d周期体到各种纳米材料,从非周期(0D)分子到一维(1D)半导体单壁纳米管,再到二维(2D)单层周期。对于每一种安排,所有结构都进行了优化。然后,利用coupled-perturbed-Hartree-Fock/Kohn-Sham方法和混合B3LYP泛函,以及在CRYSTAL17程序中实现的全电子基组(Be from的基组,Se from的基组),模拟了它们的红外和拉曼光谱。
03
计算方法
基于MOLPRO,对于分子BeSe,在多参考构型相互作用(MRCI+Q)水平上使用后Hartree-Fock方法进行电子计算,硒原子采用(27s,18p,14d,4f,3g,2h)/augc-cc-pv5z基组,铍原子采用(15s,9p,5d,4f, 3g,2h)/ augc-cc-pv5z基组。
结果与讨论
04
体块的结构以图形方式显示为图1。晶体结构为锌闪锌矿,即面心立方晶格,属于F-43m空间群,每个原始单元有一个公式单位。

图2显示了BeSe双原子分子电子态随核间分离的MRCI +Q势能曲线。
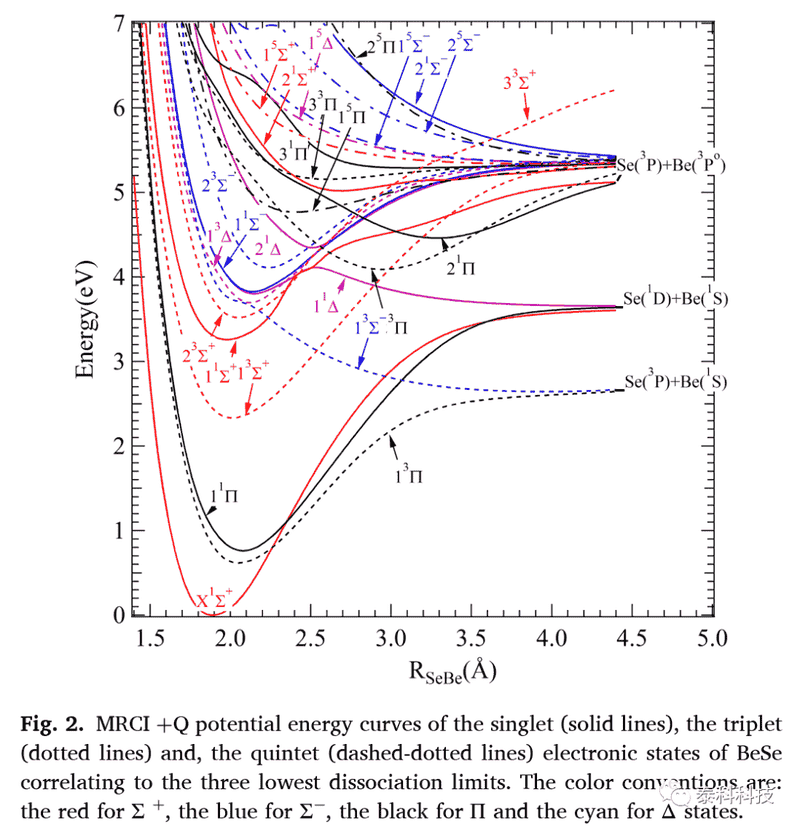
表1列出了激发能、平衡距离和三个最低电子态的主导电子构型。优化的基态平衡距离为1.88 ?,比BeSe块体的Be-Se键长至少小0.37 ?。

h-BeSe单层被发现排列在一个平面的二维六边形晶格中,其中be和Se原子被排列成蜂窝结构(见图3)。

计算得到的h-BeSe单层的红外光谱以及红外模式的原子运动如图4所示。

弛豫能δE是纳米管的优化能量与未弛豫纳米管的计算能量之差。对于两种手性,随着n的增加,这两个量都迅速减小,随着管径的增加,几乎可以忽略不计(见图5)。

在图6中,T. Larbi教授团队展示了(n,0)之字形和(n, n)扶手椅型SWBeSeNTs的n从6到48的不同值时的红外光谱。此外,图7显示了(14,0)之字形和(14,14)扶手状SWBeSeNTs的计算红外光谱,其中最大强度模态的原子位移显示在图中。

(n,0) z型和(n,n)扶手椅型SWBeSeNTs在n = 6 ~ 48时的拉曼光谱模拟如图8所示,(14,0)之字形和(14,14)扶手椅所有模式的映射如图9所示。

05
总 结
通过结合MRCI+Q和全电子DFT从头模拟,T. Larbi教授团队以不同的形式对BeSe进行了广泛的研究,从(3D)体到各种纳米排列,从零维(0D)分子到一维(1D)半导体单壁具有之字形和扶手手性的纳米管,再到二维(2D)单层,解决结构稳定性和振动特性。对于分子,在MRCI+Q方法中可以推导出关于分子最低电子态的结构和光谱的准确预测数据。从结合能的角度观察了h-BeSe的结构稳定性。在这两种手性中,松弛能、滚动能和内聚能随管径的增大而迅速减小,表现出良好的结构稳定性。它们的带隙能略大于整体带隙能,并随n的增加而略有增加,逐渐趋于h-BeSe单层的Egap值,保持了半导体性质。两种手性的BeSe纳米管的Be-Se键长与h-BeSe单层的计算值相当。它们的红外和拉曼光谱主要包含低频的径向屈曲模态,在无限长的纳米管半径下振动频率消失,而高频模态则有规律地趋向于对应单层的光学模态。
文章详情: https://doi.org/10.1016/j.surfin.2021.101087

公司简介
06
北京泰科博思科技有限公司(Beijing Tech-Box S&T Co. Ltd.)成立于2007年,是国内领先的分子模拟及虚拟仿真综合解决方案提供商。
北京泰科博思科技有限公司与国际领先的模拟软件厂商、开发团队深入合作,为高校、科研院所和企业在材料、化工、药物、生命科学、环境、人工智能及数据挖掘、虚拟仿真教学等领域提供专业的整体解决方案。用户根据需要在我们的平台上高效的进行各种模拟实验,指导实际的生产设计。
北京泰科博思科技有限公司拥有一支一流的技术服务团队和资深的专家咨询团队,以客户真正需求出发,服务客户,为客户创造价值。我们秉承“职业、敬业、担当、拼搏、合作”的企业精神,致力于用国际领先的软件产品和专业全面的技术支持服务,成为客户可信赖的合作伙伴。
2022年18期应用案例赏析-01
END

扫二维码|关注我们
了解更多咨询
来源:北京泰科博思科技有限公司
声明:本站部分文章及图片转载于互联网,内容版权归原作者所有,如本站任何资料有侵权请您尽早请联系jinwei@zod.com.cn进行处理,非常感谢!